Hypernatremia is a common electrolyte problem that is defined as a rise in serum sodium concentration to a value exceeding 145 mmol/L. [1, 2, 3] It is strictly defined as a hyperosmolar condition caused by a decrease in total body water (TBW) [4] relative to electrolyte content. Hypernatremia is a “water problem,” not a problem of sodium homeostasis.
Community-acquired hypernatremia generally occurs in elderly people who are mentally and physically impaired, often with an acute infection. Patients who develop hypernatremia during the course of hospitalization have an age distribution similar to that of the general hospital population. In both patient groups, hypernatremia is caused by impaired thirst and/or restricted access to water, often exacerbated by pathologic conditions with increased fluid loss.
The development of hyperosmolality from the water loss can lead to neuronal cell shrinkage and resultant brain injury. Loss of volume can lead to circulatory problems (eg, tachycardia, hypotension).
Acute symptomatic hypernatremia, defined as hypernatremia occurring in a documented period of less than 24 hours, should be corrected rapidly. Chronic hypernatremia (>48 h), however, should be corrected more slowly due to the risks of cerebral edema during treatment.
Pathophysiology
Hypernatremia results from a net water loss or a sodium gain, and it reflects too little water in relation to total body sodium and potassium. In a simplified view, the serum sodium concentration (Na+) can be seen as a function of the total exchangeable sodium and potassium in the body and the total body water. [5] The formula is expressed below:
Na+ = Na+ total body + K+ total body/total body water
Consequently, hypernatremia can only develop as a result of either a loss of free water or a gain of sodium or a combination of both. Hypernatremia by definition is a state of hyperosmolality, because sodium is the dominant extracellular cation and solute. [6]
The normal plasma osmolality (Posm) lies between 275 and 290 mOsm/kg and is primarily determined by the concentration of sodium salts. (Calculated plasma osmolality: 2(Na) mEq/L + serum glucose (mg/dL)/18 + BUN (mg/dL)/2.8). Regulation of the Posm and the plasma sodium concentration is mediated by changes in water intake and water excretion. This occurs via two mechanisms:
In a healthy individual, thirst and AVP release are stimulated by an increase in body fluid osmolality above a certain osmotic threshold, which is approximately 280-290 mOsm/L and is considered to be similar if not identical for both thirst and AVP release. An increased osmolality draws water from cells into the blood, thus dehydrating specific neurons in the brain that serve as osmoreceptors or “tonicity receptors.” It is postulated that the deformation of the neuron size activates these cells (thus acting like mechanoreceptors). On stimulation, they signal to other parts of the brain to initiate thirst and AVP release, resulting in increased water ingestion and urinary concentration, rapidly correcting the hypernatremic state.
Urinary concentration - AVP and the kidney
Conservation and excretion of water by the kidney depends on the normal secretion and action of AVP and is very tightly regulated. The stimulus for AVP secretion is an activation of hypothalamic osmoreceptors, which occurs when the plasma osmolality reaches a certain threshold (approximately 280 mOsm/kg). At plasma osmolalities below this threshold level, AVP secretion is suppressed to low or undetectable levels. Other afferent stimuli, such as a decrease in effective arterial blood volume, pain, nausea, anxiety, and numerous drugs, can also cause a release of AVP. [10]
AVP is synthesized in specialized magnocellular neurons whose cell bodies are located in the supraoptic and paraventricular nuclei of the hypothalamus. The prohormone is processed and transported down the axon, which terminates in the posterior pituitary gland. From there, it is secreted as active AVP hormone into the circulation in response to an appropriate stimulus (hyperosmolality, hypovolemia).
AVP binds to the V2 receptor located on the basolateral membrane of the principal cells of the renal collection ducts. The binding to this G-protein coupled receptor initiates a signal transduction cascade, leading to phosphorylation of aquaporin-2 and its translocation and insertion into the apical (luminal) membrane, creating "water channels" that enable the absorption of free water in this otherwise water-impermeable segment of the tubular system
Thirst
Thirst is the body’s mechanism to increase water consumption in response to detected deficits in body fluid. As with AVP secretion, thirst is mediated by an increase in effective plasma osmolality of only 2-3%. Thirst is thought to be mediated by osmoreceptors located in the anteroventral hypothalamus. The osmotic thirst threshold averages approximately 288-295 mOsm/kg. This mechanism is so effective that even in pathologic states in which patients are unable to concentrate their urine (diabetes insipidus) and excrete excessive amounts of urine (10-15 L/d), hypernatremia does not develop because thirst is stimulated and body fluid osmolality is maintained at the expense of profound secondary polydipsia.
Developing hypernatremia is virtually impossible if the thirst response is intact and water available. Thus, sustained hypernatremia can occur only when the thirst mechanism is impaired and water intake does not increase in response to hyperosmolality or when water ingestion is restricted.
Significant hypovolemia stimulates AVP secretion and thirst. Blood pressure decreases of 20-30% result in AVP levels many times those required for maximal antidiuresis.
Hypernatremic states can be classified as isolated water deficits (which are generally not associated with intravascular volume changes), hypotonic fluid deficits, and hypertonic sodium gain.
Regulation of brain cell volume
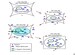
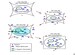
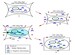
Acute hypernatremia is associated with a rapid decrease in intracellular water content and brain volume caused by an osmotic shift of free water out of the cells. Within 24 hours, electrolyte uptake into the intracellular compartment results in partial restoration of brain volume. A second phase of adaptation, characterized by an increase in intracellular organic solute content (accumulation of amino acids, polyols, and methylamines), restores brain volume to normal. Some patients complete this adaptive response in less than 48 hours. The accumulation of intracellular solutes bears the risk for cerebral edema during rehydration. The brain cell response to hypernatremia is critical. See the image below.
Figure A: Normal cell. Figure B: Cell initially responds to extracellular hypertonicity through passive osmosis of water extracellularly, resulting in cell shrinkage. Figure C: Cell actively responds to extracellular hypertonicity and cell shrinkage in order to limit water loss through transport of organic osmolytes across the cell membrane, as well as through intracellular production of these osmolytes. Figure D: Rapid correction of extracellular hypertonicity results in passive movement of water molecules into the relatively hypertonic intracellular space, causing cellular swelling, damage, and ultimately death.

Etiology
Several risk factors exist for hypernatremia. The greatest risk factor is age older than 65 years. In addition, mental or physical disability may result in impaired thirst sensation, an impaired ability to express thirst, and/or decreased access to water. [11, 12]
Hypernatremia often is the result of several concurrent factors. The most prominent is poor fluid intake. Again, developing hypernatremia is virtually impossible if the thirst response is intact and water is available. Normally, an increase in osmolality of just 1-2% stimulates thirst, as do hypovolemia and hypotension. For clinical purposes, hypernatremia can, in a simplified view, be classified on the basis of the concurrent water loss or electrolyte gain and on corresponding changes in extracellular fluid volume, as follows:
Loss of hypotonic fluid (loss of water in excess of electrolytes)
Patients who lose hypotonic fluid have a deficit in free water and electrolytes (low total body sodium and potassium) and have decreased extracellular volume. In these patients, hypovolemia may be more life-threatening than hypertonicity. When physical evidence of hypovolemia is present, fluid resuscitation with normal saline is the first step in therapy.
Renal hypotonic fluid loss results from anything that will interfere with the ability of the kidney to concentrate the urine or osmotic diuresis, such as the following:
Nonrenal hypotonic fluid loss can result from any of the following:
Pure-water deficits
Patients with pure-water deficits in the majority of cases have a normal extracellular volume with normal total body sodium and potassium. This condition most commonly develops when impaired intake is combined with increased insensible (eg, respiratory) or renal water losses.
Free-water loss will also result from an inability of the kidney to concentrate the urine. The cause of that can be either from failure of the hypothalamic-pituitary axis to synthesize or release adequate amounts of AVP (central diabetes insipidus) or a lack of responsiveness of the kidney to AVP (nephrogenic diabetes insipidus). Patients with diabetes insipidus and intact thirst mechanisms most often present with normal plasma osmolality and serum NA+, but with symptoms of polyuria and polydipsia.
Water intake less than insensible losses may result from any of the following:
Vasopressin (AVP) deficiency (diabetes insipidus)
Central diabetes insipidus [14] can be caused by any pathologic process that destroys the anatomic structures of the hypothalamic-pituitary axis involved in AVP production and secretion. Such processes include the following:
Nephrogenic diabetes insipidus (decreased responsiveness of the kidney to vasopressin)
Causes include the following:
Medications that induce nephrogenic diabetes insipidus include the following:
Medications that possibly cause nephrogenic diabetes insipidus include the following:
Adipsic diabetes insipidus (central diabetes insipidus with deficient thirst)
This is caused by a combination of damage to the osmoreceptors regulating thirst sensation and central diabetes insipidus (see above). [16] Etiologies include the following:
Gestational diabetes insipidus
In this form of diabetes insipidus, AVP is rapidly degraded by a high circulating level of oxytocinase/vasopressinase. It is a rare condition, because increased AVP secretion will compensate for the increased rate of degradation. Gestational diabetes insipidus occurs only in combination with impaired AVP production.
Hypertonic sodium gain
Patients with hypertonic sodium gain have a high total-body sodium and an extracellular volume overload (rare, mostly iatrogenic). When thirst and renal function are intact, this condition is transient. Causes include the following:
Water shift (transient)
Water shifts into muscle cells during extreme exercise or seizures because of increased intracellular osmoles). In clinical practice, a combination of the two may be present. For example, an intubated patient in the ICU develops hypernatremia due to hypertonic sodium gain caused by normal saline volume resuscitation and, in addition, increased free water excretion due to recovering renal failure and/or osmotic urea-diuresis caused by high-protein tube feeding.
Epidemiology
Although the incidence of hypernatremia on admission to the hospital has been estimated at 0.12-1.4%, a review by Tsipotis et al of 19,072 unselected hospitalized adults found that community-acquired hypernatremia occurred in 21%. Hospital-acquired hypernatremia developed in 25.9% of patients. [17]
A retrospective, single-center study from Austria, which included 981 patients, found that 2% of patients had hypernatremia on admission to the intensive care unit (ICU) and 7% developed hypernatremia during their stay in the ICU. [18] Analysis of data on 8140 patients from 12 French ICUs found that 11.1% developed mild hypernatremia and 4.2% developed moderate to severe hypernatremia 24 hours or more after ICU admission. [17]
A Canadian study of 8142 adult patients identified ICU-acquired hyponatremia in 11% of them and ICU-acquired hypernatremia in 26% of these patients. [19] The report found that the mortality rate in patients with ICU-acquired hyponatremia or hypernatremia was greater than that of study patients with normal serum sodium levels, being 28% versus 16% (P < 0.001), and 34% versus 16%, p < 0.001, respectively.
The groups most commonly affected by hypernatremia are elderly people and children. [20] Breastfeeding-associated neonatal hypernatremia has been recognized in infants ≤ 21 days of age who have lost ≥10% of birth weight. [21]
Prognosis
In patients with community-acquired hpernatremia, Tsipotis et al reported an adjusted odds ratio (OR) of 1.67 (95% confidence interval [CI], 1.38-2.01) for in-hospital mortality and 1.44 (95% CI, 1.32-1.56) for discharge to a short-/long-term care facility and an adjusted 10% (95% CI, 7-13) increase in length of stay. Patients with hospital-acquired hypernatremia had an adjusted OR of 3.17 (95% CI, 2.45-4.09) for in-hospital mortality and 1.45 (95% CI, 1.32-1.59) for discharge to a facility, and an adjusted 49% (95% CI, 44-53) increase in length of stay. [17]
Mortality rates of 30-48% have been shown in patients in ICUs who have serum sodium levels exceeding 150 mmol/L. [22, 23] A review of 256 patients presenting to a Turkish emergency department with severe hypernatremia (serum sodium >160 mmol/L) determined that the following factors were independently associated with mortality [24] :
Comparing hospital mortality rates for the patients without hypernatremia with those for cohort members with the condition, Darmon et al determined that the subdistribution hazard ratio for mortality from ICU-acquired hypernatremia was 2.03 for mild hypernatremia and 2.67 for moderate–to-severe hypernatremia. [22] However, whether the association of ICU-acquired hypernatremia with an increased risk for death reflects a direct effect of hypernatremia or is a marker for suboptimal quality of care is uncertain.
One study confirmed that maximum daily sodium amount is a significant risk factor for the development of acute kidney injury in patients with subarachnoid hemorrhage (SAH) who are receiving hypertonic saline therapy. Such therapy is often used to control intracranial hypertension and manage symptomatic hyponatremia. Of 736 patients in one study, 9% (64) developed acute kidney injury. For each 1 mEq/L increase in the running maximum daily serum sodium rate, the risk of developing acute kidney injury increased by 5.4 %, and the risk of death was more than twofold greater for patients who developed acute kidney injury. [25]
Early acquired hypernatremia in the ICU has been found to be a frequent complication in patients with severe sepsis and is associated with the volume of 0.9% saline received during the first 48 hours of admission in the ICU. In one study, of 95 patients with severe sepsis, 29 (31%) developed hypernatremia within 5 days. For every 50 ml/kg increase in 0.9% saline intake during the first 48 hours, the odds of hypernatremia increased by 1.61 times. Patients who developed hypernatremia had increased duration of mechanical ventilation and increased mortality. [26]
According to a study by Leung et al, preoperative hypernatremia is associated with increased perioperative 30-day morbidity and mortality. In their study, 20,029 patients with preoperative hypernatremia (>144 mmol/L) were compared with 888,840 patients with a normal baseline sodium (135-144 mmol/L). The odds of morbidity and mortality increased according to the severity of hypernatremia (P < 0.001 for pairwise comparison for mild [145-148 mmol/L] vs severe [>148 mmol/L] categories). Hypernatremia, versus normal baseline sodium, was associated with a greater odds for perioperative major coronary events (1.6% vs 0.7%), pneumonia (3.4% vs 1.5%), and venous thromboembolism (1.8% vs 0.9%). [27]
Treatment-resistant hypernatremia has been reported in patients with severe COVID-19. Zimmer et al found no correlation between plasma sodium concentrations and sodium input in these patients, along with elevations in plasma chloride concentrations and decreases in potassium—findings consistent with an abnormal increase in renal sodium reabsorption, possibly caused by increased angiotensin II activity secondary to virally induced downregulation of angiotensin-converting enzyme 2 (ACE2) receptors. [28] Hypernatremia in COVID-19 has been associated with the need for ventilator support, increased length of ICU stay, sepsis, altered mental status, and high mortality. [29, 30, 31]